
Genetic diversity in Vietnamese Ven dogs: A comprehensive assessment through D-loop hypervariable region 1 sequences
Do Vo Anh Khoa1* Le Cong Trieu2 Nguyen Thanh Cong3 Tran Hoang Dung4 Nguyen Thi Dieu Thuy5 Thai Ke Quan6 Dang Quoc Quan7 Nguyen Thi Thuy Trang1 Nguyen Thi Ngoc Linh8 Nguyen Huy Tuong9 Phạm Ngọc Thảo Vy9
- Vietnam National University of Forestry, Dong Nai, Vietnam
- Soc Trang Vocational College, Can Tho City, Vietnam
- Nguyen Tat Thanh University, Ho Chi Minh City, Vietnam
- Ho Chi Minh City University of Industry and Trade, Ho Chi Minh City, Vietnam
- Institute of Biology, Vietnam Academy of Science and Technology, Hanoi, Vietnam
- Saigon University, Ho Chi Minh City, Vietnam,
- University of Science, Ho Chi Minh City, Vietnam
- Can Tho University, Can Tho City, Vietnam
- Vinh Long University of Technology Education, Vinh Long Province, Vietnam
Article Information
- Date Received: 18/09/2025
- Date Revised: 07/04/2026
- Date Accepted: 08/04/2026
- Date Published Online: 27/04/2026
Copyright: © 2026 The Authors. Published by MARCIAS AUSTRALIA, 32 Champion Drive, Rosslea, Queensland 4812, Australia. This is an open access publication under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Citation: Khoa DVA, Trieu LC, Cong NT, Dung TH, Thuy NTD, Quan TK, Quan DQ, Trang NTT, Linh NTN, Tuong NH, Vy PNT (2025). Genetic diversity in Vietnamese Ven dogs: A comprehensive assessment through D-loop hypervariable region 1 sequences. Aust J Agric Vet Anim Sci (AJAVAS), 1(3), 100007
https://doi.org/10.64902/ajavas.2025.100007
Abstract
This study characterised mitochondrial genetic diversity in Vietnamese Ven dogs using a 582-bp fragment of the HV1 region from 21 individuals. A total of 32 polymorphic sites was identified, and sequence variation resolved 14 haplotypes assigned to three major haplogroups (A, B, and C). Haplogroup A was predominant, while haplotype C2 was the most frequent (19.06%). One novel haplotype (Cn) was detected at low frequency (4.67%). Genetic diversity indices indicate substantial maternal variation within the sampled population (Hd = 0.943; Pi = 0.01524), consistent with patterns reported in other Vietnamese and Southeast Asian village dogs. The haplotype network further supports the coexistence of both closely related and moderately divergent maternal lineages, although demographic inference remains limited by sample size. Overall, the findings suggest that Ven dogs represent a phenotypically defined subset embedded within a diverse regional gene pool rather than a distinct maternal lineage. This study provides a baseline HV1 dataset for Ven dogs and highlights the need for expanded sampling and genome-wide analyses to better resolve population structure, evolutionary history, and implications for conservation and breeding strategies.
Keywords:
Ven dog, HV1, D-loop, haplotype, haplogroup, genetic diversity, mitochondrial DNA
Highlights
- Fourteen distinct haplotypes grouped into three major haplogroups (A, B, C), with haplotype C2 being the most prevalent at 19.06%, were identified.
- A high genetic diversity within the sampled population, indicated by a haplotype diversity of 0.943 and nucleotide diversity of 0.01524 was found.
- A new hitherto undocumented haplotype in previous studies Cn was discovered in 4.7% of the population sampled
1.0 Introduction
Village dogs – locally referred to as “Muc,” “Ven,” and “Co”- represent long-established indigenous canine populations of Vietnam, distinct from recently introduced cosmopolitan breeds. Archaeological and ethnobiological evidence suggests that these dogs have been embedded within human settlements across the Indochinese peninsula for at least ~6,000 years, reflecting a prolonged history of cohabitation, semi-managed breeding, and ecological adaptation (Corbett, 1995). Phenotypically, Vietnamese village dogs are typically medium-sized (10–25 kg; 45–65 cm at the withers), with short coats exhibiting extensive color polymorphism, including solid and patterned forms such as brindle. This phenotypic heterogeneity likely mirrors underlying genetic diversity shaped by weak artificial selection and strong local environmental filtering. Within this heterogeneous population, the brindled phenotype – commonly referred to as the “Ven” dog – constitutes a conspicuous and geographically widespread ecotype. While “Ven” is defined primarily by coat pattern rather than strict breed status, it represents a biologically meaningful subset for population genetic inference, as phenotypic clustering in village dogs may partially track lineage structure, historical gene flow, or localized selection regimes. Consequently, Ven dogs provide an informative model for interrogating the evolutionary history and standing genetic variation of Southeast Asian village dog populations.
To date, research on Ven dogs has been dominated by phenotypic and production-oriented descriptors, including morphology, physiology, and basic reproductive traits. These studies consistently depict Ven dogs as small-to-medium-sized, resilient, and well adapted to low-input management systems, with behavioral traits suited for guarding and hunting (Trieu et al., 2018, 2019, 2020). Population-level surveys further reveal substantial within-group variability in coat pattern, ear morphology, and minor anatomical traits such as dewclaw number and tongue pigmentation. While informative, such datasets remain largely descriptive and offer limited resolution for reconstructing population history or inferring evolutionary processes. At the molecular level, preliminary candidate gene analyses have identified polymorphisms of potential functional relevance, including adjacent SNPs in the HTR1D gene that may influence amino acid composition and, by extension, behavioral phenotypes such as aggression (Trieu et al., 2020). However, these locus-specific insights do not capture genome-wide or matrilineal patterns of diversity, which are critical for understanding demographic history, maternal lineage structure, and phylogeographic relationships.
In this context, mitochondrial DNA (mtDNA), particularly the hypervariable region I (HVI) of the control region, has emerged as a powerful marker for resolving maternal genetic structure in domestic dogs. Due to its high mutation rate, lack of recombination, and maternal inheritance, mtDNA HVI enables fine-scale discrimination of haplotypes and facilitates inference of population expansion, migration, and lineage diversification. Previous studies in Vietnamese dogs – including Phu Quoc, H’Mong, and populations from the Ma River basin and southern urban regions – have consistently revealed substantial haplotype diversity and the presence of globally distributed haplogroups (A, B, C), alongside the sporadic occurrence of the rarer haplogroup E (Quan et al., 2016a,b; Tran et al., 2016; Nguyen et al., 2019; Hai et al., 2021; Bui et al., 2021). These findings position Vietnam within the broader Southeast Asian domestication landscape, a region increasingly recognized as a major reservoir of early canine genetic diversity.
Despite these advances, the maternal genetic architecture of Ven dogs remains poorly characterised. Specifically, it is unclear whether this phenotypically defined group represents a random subset of the broader village dog gene pool or retains distinct haplotypic signatures indicative of structured ancestry, localized selection, or demographic isolation. Addressing this gap requires a shift from descriptive phenotyping to sequence-based population genetic analysis. The present study therefore aims to characterize the genetic diversity of Vietnamese Ven dogs using mtDNA HVI sequences, with an emphasis on haplotype diversity, nucleotide variation, and phylogenetic affiliation. By integrating newly generated sequence data with published datasets from other Vietnamese dog populations, we seek to (i) resolve the maternal lineage composition of Ven dogs, (ii) assess their position within regional haplogroup structure, and (iii) provide a foundational dataset for subsequent genome-wide and coalescent-based analyses of village dog evolution in Vietnam.
2.0 Materials and methods
2.1. Sample collection and total DNA extraction
A total of 21 putatively unrelated Vietnamese Ven dogs were sampled across multiple provinces of the Mekong Delta, including Ca Mau (n = 4), Soc Trang (n = 3), Hau Giang (n = 3), An Giang (n = 4), Can Tho (n = 3), and Vinh Long (n = 4) (Trieu, 2024). Unrelatedness was approximated through owner interviews to minimize sampling of closely related individuals, a common constraint in village dog populations lacking formal pedigree records. The sampled dogs were distributed across communities representing diverse ethnic backgrounds (Kinh, Khmer, and Hoa), thereby capturing a degree of socio-ecological heterogeneity that may influence gene flow and breeding structure.
All individuals were phenotypically classified as Ven dogs based on the presence of brindle coat patterns, typically expressed as combinations of black, yellow, and white pigmentation. Each sample was assigned a unique identifier (ST1–ST21). To reduce the inclusion of recently introgressed or non-native lineages, a morphological screening protocol was applied during sampling, excluding individuals exhibiting diagnostic traits of recognized foreign breeds (Figure 1). While morphology-based filtering cannot fully eliminate cryptic admixture, it provides a pragmatic first-pass control in field-based population genetic studies of free-breeding dogs.
Genomic DNA was extracted from hair samples using a standardized protocol adapted from Quan et al., (2016c) Quan et al. (2016c), optimized for low-input keratinized tissues. DNA quality and concentration were assessed via spectrophotometric analysis (NanoDrop™ One/OneC, Thermo Scientific™), ensuring adequate purity for downstream applications. All DNA extracts were subsequently normalized to a working concentration of 20 ng/µL to standardize template input for amplification of the mitochondrial hypervariable region I (HVI).




Figure 1. Some primary dog coat colours with the brindle phenotype (top) and characteristics of the brindle ridge dog (bottom)
2.2. Sequencing
The HV1 region gene sequence (582 bp) was amplified by PCR using a specific primer pair (15412F: CCACTATCAGCACCCAAAG, and 16625R: AGACTACGAGACCAAATGC) (Gundry et al., 2007). This locus was selected due to its elevated mutation rate and established utility in resolving fine-scale maternal lineage structure in domestic dogs. PCR amplifications were performed in a total reaction volume of 25 µL, containing 1× MyTaq™ Red Mix (Bioline), 0.2 µM of each primer, approximately 20 ng of template DNA, and nuclease-free water. Thermal cycling was conducted under the following conditions: an initial denaturation at 95°C for 5 min; followed by 35 cycles of denaturation at 95°C for 1 min, annealing at 52°C for 30 s, and extension at 72°C for 1 min; with a final extension at 72°C for 5 min. These parameters were optimized to ensure robust amplification of mitochondrial templates while minimizing nonspecific products. Amplification success was verified by electrophoresis on 1.5% agarose gels stained with GelRed®, with fragment size assessed against a standard DNA ladder. PCR products exhibiting single, clear bands of the expected size were purified to remove residual primers and dNTPs prior to sequencing. Purified amplicons were then subjected to bidirectional Sanger sequencing at a commercial facility (1st BASE, Malaysia). This targeted sequencing approach enables high-confidence base calling across the HVI region, providing the resolution necessary for downstream haplotype identification, polymorphism detection, and phylogenetic inference of maternal lineages.
2.3. Data analysis
The chromatograms obtained from sequencing were analyzed using FinchTV 1.4.0. Any inconsistencies observed between the forward and reverse sequences were resolved manually. Subsequently, all sequences were aligned with a reference sequence (GenBank no: U96639.2) using MEGA 11 (Tamura et al., 2021) and trimmed to generate sequences of 582 bp. Nucleotide positions were annotated according to Pereira’s scheme (Pereira et al., 2004). These 582 bp sequences were then subjected to Haplotype Identifier for haplotyping (Thai et al., 2017). Any sequence exhibiting new mutations was designated with the suffix “n” to indicate novelty, for instance, A1n, C2n. For a comprehensive understanding of haplotype relationships, a haplotype medium-joining network was constructed using Network 10.2.0.0 (Bandelt et al., 1999) and DnaSP version 6.11 (Rozas et al., 2017). Genetic diversity indices such as haplotype diversity (Hd), nucleotide diversity (Pi), and average number of distinct nucleotides (K) were computed using Arlequin (Excoffier et al., 2010).
3.0 Results
3.1. Sequence variation in the 582-bp HV1 fragment
Analysis of the 582-bp mitochondrial control region (HV1) across 21 Vietnamese Ven dogs revealed a total of 32 polymorphic sites (Table 1), indicating a moderate-to-high level of maternal sequence variability within this population. The mutational spectrum was strongly biased toward transitions (29/32 sites), with only a single transversion and two insertion–deletion (indel) events detected. This pronounced transition bias is a well-documented feature of mitochondrial control regions and supports the authenticity of the observed variation, as it conforms to established patterns of mtDNA molecular evolution rather than suggesting systematic sequencing error.
3.3. Haplotype network structure and mutational distances
The median-joining network (Figure 2) resolves the genealogical relationships among the 14 identified haplotypes and clearly delineates their partitioning into three major mitochondrial haplogroups: A, B, and C. This structure is consistent with the haplogroup assignments derived from diagnostic polymorphisms and reinforces the presence of multiple maternal lineages within the Ven dog population.
Across the network, mutational distances among haplotypes vary substantially, ranging from one to two substitutions within localized clusters to as many as ~12 mutational steps between more distantly related nodes. This broad spectrum of divergence indicates the coexistence of both recently differentiated haplotypes and more deeply separated maternal lineages. Within haplogroups, several haplotypes form compact clusters characterized by short mutational branches, suggestive of recent diversification or shared ancestry. In contrast, longer branches connecting haplogroups or peripheral haplotypes likely reflect older coalescent events and deeper phylogenetic separation.
Notably, the network does not exhibit a strongly resolved star-like topology centred on a single high-frequency haplotype. While this may suggest the absence of a recent, rapid demographic expansion signal, such an inference remains tentative. Median-joining networks are inherently sensitive to sampling density, and the limited sample size (n = 21) increases the likelihood that intermediate or ancestral haplotypes are missing. This can artificially elongate branches, inflate apparent mutational distances, and obscure central nodes that would otherwise indicate expansion dynamics.
Accordingly, the network is interpreted here as a qualitative representation of maternal lineage diversity rather than a definitive reconstruction of demographic history. The observed topology robustly supports the coexistence of multiple, partially differentiated maternal lineages within Vietnamese Ven dogs, but further inference regarding population expansion, bottlenecks, or lineage turnover will require larger sample sizes and formal model-based approaches (e.g., Bayesian skyline analyses or coalescent simulations).
Sequence divergence among haplotypes was non-uniform, reflecting a mixture of closely related and more divergent maternal lineages within the sampled population. Relative to the reference sequence (GenBank: U96639.2), certain haplotypes – most notably B1, B6, and Cn – exhibited a higher number of mutational differences (Table 1), suggesting deeper genealogical separation or potential affiliation with distinct haplogroup sublineages. Conversely, other haplotypes differed by only a small number of substitutions, consistent with recent divergence or shared ancestry within localized maternal clusters.
Importantly, several polymorphic sites were shared across multiple haplotypes, forming recurring mutational motifs. Such patterns are indicative of lineage-specific signatures rather than independent mutational events, and they likely reflect the hierarchical structure of mtDNA haplogroups. This observation reinforces the utility of HV1 variation for resolving phylogenetic relationships at the intra-population level.
Indel variation, while limited in frequency, warrants careful interpretation due to its sensitivity to alignment parameters and sequencing artifacts. In the present dataset, the Cn haplotype uniquely harbors an indel event not observed in other sequences (Table 1). Given the potential for indel miscalling in homopolymeric or repetitive regions of the control region, this feature is treated here as a provisional diagnostic marker of the Cn haplotype. Rigorous validation – particularly through chromatogram inspection and, ideally, replicate sequencing – is recommended to confirm the stability and reproducibility of this indel signal. Overall, the observed pattern of sequence variation suggests that Vietnamese Ven dogs harbor a heterogeneous assemblage of maternal lineages, combining both shallow and deep divergence. This provides a necessary foundation for subsequent haplotype-based and phylogeographic analyses aimed at reconstructing population history and lineage connectivity.
Table 1. Polymorphic sites in the 582 bp HV1 region sequence*
| Sample | 15464 | 15484 | 15508 | 15526 | 15553 | 15557 | 15595 | 15611 | 15612 | 15613 | 15620 | 15627 | 15630 | 15632 | 15639 |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Ref. | C | – | A | C | C | A | T | C | T | T | A | T | A | C | C |
| A8 | . | – | . | . | . | . | . | . | . | . | . | . | G | . | . |
| A9 | . | – | . | . | . | . | . | . | . | . | . | . | G | . | . |
| A11 | . | – | . | . | . | . | . | . | . | . | . | . | . | . | . |
| A17 | . | – | . | . | . | . | . | . | . | . | . | C | G | . | . |
| A18 | . | – | . | . | . | . | . | . | . | . | . | . | . | . | . |
| A65 | . | – | . | . | . | . | . | . | . | . | . | . | . | . | . |
| A73 | . | – | . | . | . | . | . | . | . | . | . | . | G | . | . |
| A121 | . | – | . | . | . | . | . | . | . | . | . | . | G | . | . |
| A132 | T | – | G | . | . | . | C | . | . | . | . | . | G | T | . |
| A223 | . | – | . | . | . | . | . | . | . | . | . | . | G | . | . |
| B1 | . | – | . | . | T | . | . | T | . | C | . | . | . | . | T |
| B6 | . | C | . | . | T | . | . | . | . | C | . | . | . | . | T |
| C2 | . | – | . | T | T | . | . | . | C | . | . | . | . | . | . |
| Cn | . | – | . | T | T | G | . | . | C | . | G | . | . | . | . |
| Sample | 15464 | 15484 | 15508 | 15526 | 15553 | 15557 | 15595 | 15611 | 15612 | 15613 | 15620 | 15627 | 15630 | 15632 | 15639 |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Ref. | C | – | A | C | C | A | T | C | T | T | A | T | A | C | C |
| A8 | . | – | . | . | . | . | . | . | . | . | . | . | G | . | . |
| A9 | . | – | . | . | . | . | . | . | . | . | . | . | G | . | . |
| A11 | . | – | . | . | . | . | . | . | . | . | . | . | . | . | . |
| A17 | . | – | . | . | . | . | . | . | . | . | . | C | G | . | . |
| A18 | . | – | . | . | . | . | . | . | . | . | . | . | . | . | . |
| A65 | . | – | . | . | . | . | . | . | . | . | . | . | . | . | . |
| A73 | . | – | . | . | . | . | . | . | . | . | . | . | G | . | . |
| A121 | . | – | . | . | . | . | . | . | . | . | . | . | G | . | . |
| A132 | T | – | G | . | . | . | C | . | . | . | . | . | G | T | . |
| A223 | . | – | . | . | . | . | . | . | . | . | . | . | G | . | . |
| B1 | . | – | . | . | T | . | . | T | . | C | . | . | . | . | T |
| B6 | . | C | . | . | T | . | . | . | . | C | . | . | . | . | T |
| C2 | . | – | . | T | T | . | . | . | C | . | . | . | . | . | . |
| Cn | . | – | . | T | T | G | . | . | C | . | G | . | . | . | . |
*The numbers in the vertical column describe the nucleotide positions. The sign: (.) represents a nucleotide that matches the reference sequence; (-) represents a nucleotide deletion site. Ref: represents the reference sequence (Imes et al., 2012).
3.2. Haplotype compositions and haplogroup structure
Applying the haplotype classification framework described above, a total of 14 distinct haplotypes were identified among the 21 HV1 sequences obtained from Vietnamese Ven dogs, which were subsequently assigned to three major mitochondrial haplogroups: A, B, and C (Table 2). This tripartite structure is consistent with the dominant maternal lineages reported globally in domestic dogs and widely observed across Southeast Asia. Haplogroup A was the most prevalent, encompassing 10 haplotypes distributed across 12 individuals (57.1%), thereby representing the principal maternal lineage within the sampled population. In contrast, haplogroup B comprised two haplotypes represented by four individuals (19.0%), while haplogroup C also included two haplotypes but accounted for five individuals (23.8%). The co-occurrence of these three haplogroups within a relatively small sample set underscores the absence of maternal lineage homogeneity and suggests that Ven dogs do not derive from a single, isolated maternal ancestry. Rather, they appear to represent a composite population shaped by multiple lineage contributions.
At finer resolution, haplotype frequencies were highly skewed toward low-frequency variants, with the majority of haplotypes observed as singletons (1/21; ~4.8%). Such a singleton-rich distribution is characteristic of populations with either high standing genetic diversity or incomplete sampling of intermediate lineages. In the context of free-breeding village dogs, this pattern is plausibly driven by a combination of weak artificial selection, extensive gene flow, and large effective population sizes, all of which facilitate the persistence of rare maternal lineages. However, given the modest sample size, caution is warranted in extrapolating haplotype frequencies to the broader Ven dog population. The current dataset likely underrepresents the full spectrum of haplotypic diversity, and the observed frequency distribution should therefore be interpreted as descriptive rather than inferential. From a coalescent perspective, the high proportion of low-frequency haplotypes may also be compatible with demographic expansion or long-term population stability, hypotheses that require formal testing using larger datasets and model-based approaches.
Table 2. The haplotypes identified in 21 individual Ven dogs
| Haplogroup | Haplotype | Sample Name | Quantity / % |
|---|---|---|---|
| A | A8 | ST19 | 1 (4.67%) |
| A9 | ST12 | 1 (4.67%) | |
| A11 | ST6, ST8, ST16 | 3 (14.29%) | |
| A17 | ST13 | 1 (4.67%) | |
| A18 | ST2 | 1 (4.67%) | |
| A65 | ST1 | 1 (4.67%) | |
| A73 | ST4 | 1 (4.67%) | |
| A121 | ST20 | 1 (4.67%) | |
| A132 | ST5 | 1 (4.67%) | |
| A223 | ST15 | 1 (4.67%) | |
| B | B1 | ST3, ST10, ST11 | 3 (14.29%) |
| B6 | ST17 | 1 (4.67%) | |
| C | C2 | ST9, ST14, ST18, ST121 | 4 (19.06%) |
| Cn | ST7 | 1 (4.67%) |
3.3. Haplotype network structure and mutational distances
The median-joining network (Figure 2) resolves the genealogical relationships among the 14 identified haplotypes and clearly delineates their partitioning into three major mitochondrial haplogroups: A, B, and C. This structure is consistent with the haplogroup assignments derived from diagnostic polymorphisms and reinforces the presence of multiple maternal lineages within the Ven dog population.
Across the network, mutational distances among haplotypes vary substantially, ranging from one to two substitutions within localized clusters to as many as ~12 mutational steps between more distantly related nodes. This broad spectrum of divergence indicates the coexistence of both recently differentiated haplotypes and more deeply separated maternal lineages. Within haplogroups, several haplotypes form compact clusters characterized by short mutational branches, suggestive of recent diversification or shared ancestry. In contrast, longer branches connecting haplogroups or peripheral haplotypes likely reflect older coalescent events and deeper phylogenetic separation.
Notably, the network does not exhibit a strongly resolved star-like topology centred on a single high-frequency haplotype. While this may suggest the absence of a recent, rapid demographic expansion signal, such an inference remains tentative. Median-joining networks are inherently sensitive to sampling density, and the limited sample size (n = 21) increases the likelihood that intermediate or ancestral haplotypes are missing. This can artificially elongate branches, inflate apparent mutational distances, and obscure central nodes that would otherwise indicate expansion dynamics.
Accordingly, the network is interpreted here as a qualitative representation of maternal lineage diversity rather than a definitive reconstruction of demographic history. The observed topology robustly supports the coexistence of multiple, partially differentiated maternal lineages within Vietnamese Ven dogs, but further inference regarding population expansion, bottlenecks, or lineage turnover will require larger sample sizes and formal model-based approaches (e.g., Bayesian skyline analyses or coalescent simulations).
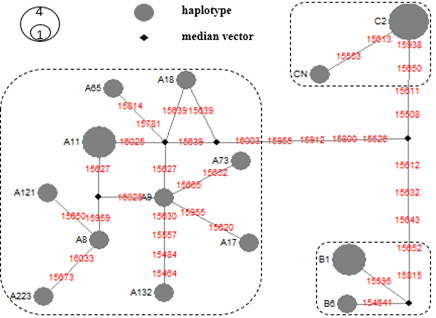
Figure 2. The haplotype network (median-joining network).
The haplotype network (median-joining network) was constructed based on polymorphic positions in the HV1 sequence region, showing the relationship of 14 haplotypes belonging to 3 haplogroups (A, B, C) found in 21 brindle coat individuals. The size of the haplotype node represents how often individuals have the same corresponding haplotype. The black circles (median vectors) represent intermediate haplotypes that have not been recorded in the study. The digits on the line connecting the haplotypes represent the mutation sites between the two haplotypes corresponding to the location of the HV1 sequence on the complete mitochondrial genome (Kim et al., 2001).
3.4. Genetic diversity indices and comparative interpretation across populations
Summary statistics of mitochondrial diversity derived from the 582-bp HV1 dataset are presented in Table 3. The Ven dog population exhibits a high level of haplotype diversity (Hd = 0.943), indicating a strong probability that any two randomly sampled individuals possess distinct maternal haplotypes. Such elevated Hd values are characteristic of free-breeding village dog populations, where weak artificial selection and extensive gene flow facilitate the maintenance of multiple coexisting lineages.
Nucleotide diversity (Pi) and the mean number of pairwise nucleotide differences (K) provide complementary measures of sequence divergence across the analyzed fragment. Together, these indices demonstrate that genetic variation is distributed across multiple polymorphic sites rather than being confined to a small number of hypervariable positions. This pattern is consistent with the coexistence of both shallow and moderately deep maternal divergences within the population, as also reflected in the haplotype network structure.
When considered in a broader regional context, the diversity estimates for Ven dogs fall within the range reported for other Vietnamese indigenous dog populations, including Phu Quoc ridgebacks, H’Mong bobtailed dogs, and village dogs from the Ma River basin and southern urban regions (Table 3). This concordance suggests that Ven dogs are not genetically depauperate despite being defined by a specific coat phenotype; rather, they retain a level of maternal diversity comparable to other recognized native populations. From a phylogeographic perspective, this supports the view that phenotypic classification (e.g., brindle patterning) does not necessarily correspond to discrete genetic lineages but may instead be superimposed upon a shared and heterogeneous maternal gene pool.
Importantly, while high Hd combined with moderate Pi is often interpreted as a signal of population expansion following a period of low effective population size, such demographic inferences remain tentative in the absence of formal neutrality tests or coalescent-based modeling. Given the limited sample size, the present estimates should therefore be interpreted as indicative of substantial standing genetic variation rather than definitive evidence of specific demographic scenarios.
Table 3. Haplotype diversity index, nucleotide diversity and average number of distinct nucleotides in some dog populations in Vietnam and around the world.
| Group / Breed of Dog | Haplotype Diversity (Hd) | Nucleotide Diversity (Pi) | Average Number of Distinct Nucleotides (K) | Reference |
|---|---|---|---|---|
| Ven | 0.943 | 0.01524 | 8.85238 | Khoa et al. (2025) |
| Phu Quoc ridgeback | 0.9042 | 0.014588 | 8.519596 | GenBank code MG793253–MG793352 |
| H’Mong bobtailed | 0.952 | 0.02299 | 13.31053 | Hai et al. (2021) |
| Ma River village | 0.969 | 0.00912 | 5.456 | Bui et al. (2021) |
| House dogs in Ho Chi Minh City | 0.8814 | 0.014035 | 8.182424 | Quan et al. (2016b) |
| Thai | 0.9493 | 0.009599 | 5.595971 | Pang et al. (2009); Savolainen et al. (2002) |
| Jindo | 0.7308 | 0.006645 | 3.867199 | |
| Pungsan | 0.9064 | 0.011367 | 6.615385 | |
| German Shepherd | 0.6842 | 0.008681 | 5.052632 | |
| Portuguese Shepherd | 0.4841 | 0.008291 | 4.825397 | Van Asch et al. (2005) |
| Maltese | 0.8046 | 0.011758 | 6.855172 | Takahasi et al. (2002) |
| Kangal | 0.8407 | 0.015272 | 8.888482 | Excoffier et al. (2010), Imes et al. (2012), Savolainen et al. (2002), Koban et al. (2009) |
| Tibetan Mastiff | 0.8063 | 0.006645 | 3.867389 | Ren et al. (2017) |
| Shiba | 0.8161 | 0.012221 | 7.112644 | Excoffier et al. (2010), Imes et al. (2012), Savolainen et al. (2002), Okumura et al. (1996) |
Table 4 demonstrates the diversity estimates of Vietnamese Ven dogs within a broader comparative framework, incorporating both indigenous Vietnamese populations and representative global dog breeds. The observed values for haplotype diversity (Hd = 0.943) and nucleotide diversity (Pi = 0.01524) place the Ven dog dataset toward the upper range of reported variation, consistent with patterns typically associated with free-breeding village dog populations in Southeast Asia. Notably, the magnitude of diversity in Ven dogs is comparable to that reported for other Vietnamese populations, such as Phu Quoc ridgebacks and H’Mong bobtailed dogs, as well as certain non-Vietnamese populations with historically large or admixed gene pools. This reinforces the interpretation that Ven dogs retain substantial maternal genetic variation and are not genetically constrained despite being defined by a specific coat phenotype. Instead, their diversity appears to reflect a shared regional gene pool shaped by long-term gene flow and limited breeding control. However, these cross-population comparisons must be interpreted with methodological caution. The datasets included in Table 4 differ considerably in sample size, geographic coverage, and, in some cases, sequence length and analytical pipelines. Such heterogeneity can introduce bias into diversity estimates, particularly inflating haplotype diversity in smaller samples due to an overrepresentation of rare variants. As a result, the relatively high diversity observed in Ven dogs should be regarded as a contextual benchmark rather than definitive evidence of greater genetic diversity relative to other populations. Accordingly, in the revised interpretative framework, we emphasize the sensitivity of diversity metrics to sampling effects and advocate for more standardized comparative approaches. Specifically, rarefaction-based normalization, increased sample sizes, and broader geographic representation will be essential to validate the robustness of these patterns. Integration with coalescent-based inference and genome-wide data will further enable rigorous testing of whether the elevated diversity observed here reflects underlying demographic processes or sampling artifacts.
Table 4. Indices of haplotype and nucleotide diversity in some dog populations in Vietnam and around the world.
| Group / Breed of Dog | Haplotype Diversity (Hd) | Nucleotide Diversity (Pi) | Average Number of Distinct Nucleotides (K) | Reference |
|---|---|---|---|---|
| Ven | 0,943 | 0.01524 | 8.85238 | Khoa et al. (2025) |
| Phu Quoc ridgeback | 0.9042 | 0.014588 | 8,519596 | Genbank code MG793253–MG793352 |
| H’Mong bobtailed | 952 | 0.02299 | 13,31053 | Hai et al. (2021) |
| Ma River village | 0.969 | 0.00912 | 5,456 | Bui et al. (2021) |
| House dogs in Ho Chi Minh City | 0.8814 | 0.014035 | 8,182424 | Quan et al. (2016b) |
| Thai | 0.9493 | 0.009599 | 5.595971 | Pang et al. (2009); Savolainen et al. (2002) |
| Jindo | 0.7308 | 0.006645 | 3,867199 | |
| Pungsan | 0.9064 | 0.011367 | 6,615385 | |
| German Shepherd | 0.6842 | 0,008681 | 5.052632 | |
| Portuguese Shepherd | 0.4841 | 0,008291 | 4,825397 | Van Asch et al. (2005) |
| Maltese | 0.8046 | 0.011758 | 6,855172 | Takahasi et al. (2002) |
| Kangal | 0.8407 | 0.015272 | 8,888482 | Excoffier et al. (2010), Imes et al. (2012), Savolainen et al. (2002), Koban et al. (2009) |
| Tibetan Mastiff | 0.8063 | 0.006645 | 3.867389 | Ren et al. (2017) |
| Shiba | 0.8161 | 0.012221 | 7,112644 | Excoffier et al. (2010), Imes et al. (2012), Savolainen et al. (2002), Okumura et al. (1996) |
4.0 Discussion
4.1. Scope of inference: HV1 as a matrilineal marker
This study provides a first-pass, sequence-based characterization of maternal genetic diversity in Vietnamese Vện dogs using a 582-bp fragment of the mitochondrial HV1 region. The identification of 14 haplotypes across 21 individuals, distributed among three major haplogroups (A, B, and C), demonstrates that even a modest sampling captures substantial matrilineal heterogeneity. However, the interpretative scope of these findings is inherently constrained by the uniparental inheritance of mtDNA. HV1 variation reflects only maternal lineage history and, by design, cannot resolve biparental genomic structure, male-mediated gene flow, or patterns of admixture that are pervasive in free-breeding dog populations. Thus, the present results should be understood as a lineage-specific projection of diversity rather than a comprehensive representation of the genomic architecture of Ven dogs.
4.2. Maternal haplogroups in regional phylogeographic context
The detection of haplogroups A, B, and C aligns closely with prior studies of Vietnamese and broader Southeast Asian dog populations, in which these lineages consistently dominate the mitochondrial landscape (Quan et al., 2016a, Quan et al., 2016b; Tran et al., 2016; Nguyen et al., 2019; Hai et al., 2021; Bui et al., 2021). From a phylogeographic perspective, this concordance reinforces the interpretation that Ven dogs are embedded within a shared regional gene pool rather than representing a distinct or isolated maternal lineage. Haplogroup A, in particular, has been widely associated with East and Southeast Asian origins and often exhibits high internal diversity, while haplogroups B and C contribute additional layers of lineage complexity. The presence of all three haplogroups within the Ven dataset therefore supports a model of long-term lineage coexistence and gene flow, consistent with the semi-managed, low-selection breeding systems typical of village dogs. Importantly, this finding also underscores that phenotypic classification – such as brindle coat patterning – probably does not map cleanly onto mitochondrial lineage boundaries.
4.3. High haplotype diversity under sampling constraints
The high haplotype diversity (Hd = 0.943) observed in this study indicates that the sampled individuals harbor a wide array of distinct maternal lineages. In population genetic terms, such values are typically associated with large effective population sizes and/or historical admixture. However, the interpretation of Hd must be tempered by sampling considerations. With n = 21, the dataset is particularly sensitive to the presence of singleton haplotypes, which constitute a substantial fraction of the observed diversity (~4.8% per haplotype). Under expanded sampling, some of these singletons may resolve into low-frequency clusters, while additional haplotypes are likely to be discovered. Consequently, the current estimates are best viewed as evidence of high standing diversity within the sampled subset rather than precise estimators of population-level parameters. From a coalescent standpoint, the data are compatible with multiple demographic scenarios, including population expansion or long-term stability, but do not independently discriminate among them.
4.4. Network topology: Structure without overinterpretation
The median-joining network provides a useful visualization of haplotypic relationships, revealing both compact clusters and more distantly related nodes distributed across haplogroups A-C. The observed range of mutational distances supports the coexistence of both recent and more ancient maternal lineages within the population. However, network topology is inherently sensitive to sampling density. Incompletely sampled populations tend to produce fragmented networks with elongated branches, as intermediate haplotypes remain unsampled. In this context, the absence of a clear star-like expansion pattern should not be interpreted as evidence against recent demographic growth. Rather, the network should be treated as descriptive of lineage diversity and connectivity, without extending to formal demographic inference. Robust conclusions regarding population history will require integration with model-based approaches, such as Bayesian skyline analyses or approximate Bayesian computation.
4.5. Mutation spectrum and the role of indels
The predominance of transition substitutions among the 32 polymorphic sites is fully consistent with established mutational biases in mitochondrial control regions, lending confidence to the biological validity of the dataset. Indel variation, although limited, provides additional discriminatory power at the haplotype level, most notably in the case of the Cn haplotype. However, indels in HV1 are often associated with homopolymeric tracts and alignment ambiguity, making them particularly susceptible to sequencing and annotation artifacts. For this reason, indel positions should be treated as high-priority quality-control loci, requiring careful chromatogram validation and consistent alignment protocols. When rigorously verified, such variants can serve as informative markers of lineage differentiation; when not, they risk inflating apparent diversity.
4.6. Interpreting comparative diversity: benchmarking versus inference
The comparative framework presented in Table 4 situates Ven dogs within the broader landscape of canine mitochondrial diversity, where they fall within the upper range of reported values. While this is consistent with expectations for Southeast Asian village dogs, such comparisons are inherently sensitive to differences in sample size and study design. Diversity indices such as Hd and haplotype richness are known to increase with sampling effort until asymptotic levels are reached. Therefore, Table 4 should be interpreted as a contextual benchmark rather than a basis for ranking populations. Methodologically, future work would benefit from the application of rarefaction-based approaches and haplotype accumulation curves to standardize comparisons across datasets. Such approaches, combined with expanded sampling, will be essential for determining whether the high diversity observed here reflects underlying biological patterns or sampling effects.
4.7. Limitations and directions for future research
The principal limitations of this study arise from sample size and sampling design. Household-based sampling, while practical, may introduce hidden relatedness and spatial clustering, potentially biasing haplotype frequency estimates. Increasing the sample size (ideally to >100 individuals) and implementing geographically stratified sampling would substantially improve the precision and representativeness of diversity estimates. Equally important is the transition from single-locus to genome-wide inference. Because mtDNA captures only maternal inheritance, it cannot resolve the genetic architecture underlying phenotypic traits such as coat patterning. Future studies should therefore incorporate nuclear markers, including candidate genes associated with pigmentation as well as genome-wide SNP datasets, to disentangle the relationship between phenotype and genotype and to quantify admixture and population structure more comprehensively.
5.0 Conclusion
This study provides a sequence-based characterization of mitochondrial HV1 diversity in Vietnamese Ven dogs, based on a 582-bp fragment analyzed across 21 individuals. A total of 32 polymorphic sites were identified, with a mutation spectrum dominated by transitions, consistent with canonical patterns of mtDNA evolution. The dataset resolved 14 distinct haplotypes assigned to three major haplogroups (A, B, and C), including one previously unreported haplotype (Cn). The coexistence of multiple haplogroups and the high haplotype diversity (Hd = 0.943) indicate that Ven dogs harbor a heterogeneous assemblage of maternal lineages rather than a single, lineage-restricted origin. This pattern is congruent with prior studies of Vietnamese and Southeast Asian village dogs, supporting the view that Ven dogs are embedded within a broader, regionally shared maternal gene pool. The absence of haplogroups D and E further aligns with established phylogeographic distributions, where these lineages are either geographically restricted or occur at low frequencies outside specific regions. The median-joining network corroborates this interpretation by revealing both closely related and more divergent haplotypes distributed across haplogroups, although its structure remains descriptive given the limited sample size. Accordingly, the present findings should be interpreted as evidence of substantial standing maternal diversity within the sampled population, rather than as definitive inference of demographic history. Overall, this study establishes a foundational HV1 dataset for Ven dogs and reinforces the importance of Vietnamese village dogs as reservoirs of genetic diversity. Future work integrating larger, geographically stratified sampling and genome-wide markers will be essential to resolve population structure, test demographic hypotheses, and evaluate the genetic basis of phenotypic traits such as coat patterning.
Author Contributions: Conceptualisation: Do Vo Anh Khoa, Nguyen Thi Dieu Thuy; Methodology: Do Vo Anh Khoa, Tran Hoang Dung, Nguyen Thanh Cong; Software: Thai Ke Quan, Nguyen Thanh Cong; Validation: Do Vo Anh Khoa, Le Cong Trieu, Tran Hoang Dung; Formal analysis: Nguyen Thanh Cong, Tran Hoang Dung; Investigation: Do Vo Anh Khoa, Le Cong Trieu, Nguyen Huy Tuong; Resources: Do Vo Anh Khoa, Le Cong Trieu; Data Curation: Tran Hoang Dung, Nguyen Thanh Cong, Thai Ke Quan; Writing-Original Draft Preparation: Le Cong Trieu, Nguyen Thanh Cong, Thai Ke Quan, Nguyen Huy Tuong, Nguyen Thi Thuy Trang, Nguyen Thi Ngoc Linh, Pham Ngoc Thao Vy; Writing—Review and Editing: Do Vo Anh Khoa, Tran Hoang Dung, Nguyen Thi Dieu Thuy. All authors have read and agreed to the published version of the manuscript.
Funding: This research received no external funding.
Ethics Approval Statement: All research procedures were conducted in compliance with Article 72 of the Law on Livestock Production (Law 32/2018/QH14) regarding humane treatment and animal welfare and followed the National Regulation (TCVN 12448:2018) on animal welfare management. Verbal informed consent was obtained from all the Ven dog owners involved in the study as all the dog hair samples were collected with the help of their owners.
Data Availability Statement: All the relevant data that support the findings of this study are available from the corresponding author upon reasonable request.
Acknowledgments: This article was presented at the 6th National Conference on Animal and Veterinary Sciences (AVS2025, Vietnam) held by Thai Nguyen University of Agriculture and Forestry from November 6-8th, 2025.
Conflicts of Interest: The authors declare no conflicts of interest.
Artificial Intelligence: AI was not used for this original research article.
References
Bandelt HJ, Forster P, Röhl A. 1999. Median-joining networks for inferring intraspecific phylogenies. Molecular Biology and Evolution, 16(1), 37–48. https://doi.org/10.1093/oxfordjournals.molbev.a026036
Bui XP, Pham TH, Tran HC, Phung TT, Ngo QD, Dam QT, Vu DD. 2021. Evaluation of genetic diversity and origin of Song Ma village dogs in Vietnam. Biomedical and Biotechnology Research Journal, 5(4), 412–419. https://doi.org/10.4103/bbrj.bbrj_202_21
Corbett LK. 1995. The Dingo in Australia and Asia. Sydney: University of New South Wales Press, 191–196. https://catalogue.nla.gov.au/catalog/2006786
Excoffier L, Lischer HEL. 2010. Arlequin suite Ver 3.5: A new series of programs to perform population genetics analyses under Linux and Windows. Molecular Ecology Resources, 10(3), 564–567. https://doi.org/10.1111/j.1755-0998.2010.02847.x
Gundry RL, Allard MW, Moretti TR, Honeycutt RL, Wilson MR, Monson KL, Foran DR. 2007. Mitochondrial DNA analysis of the domestic dog: control region variation within and among breeds. Journal of Forensic Sciences, 52(3), 562–572. https://doi.org/10.1111/j.1556-4029.2007.00425.x
Hai PT, Phuong BX, Coi TH, Tung PT, Duc NQ, Khang NM, Duy VD. 2021. Genetic diversity of H’mong short tail dog based on sequencing of the D-loop hypervariable-1 region (HV1). Vietnam Journal of Biotechnology, 19(2), 245–257. https://doi.org/10.15625/1811-4989/14858
Imes DL, Wictum EJ, Allard MW, Sacks BN. 2012. Identification of single nucleotide polymorphisms within the mtDNA genome of the domestic dog to discriminate individuals with common HVI haplotypes. Forensic Science International: Genetics, 6(5), 630–639. https://doi.org/10.1016/j.fsigen.2012.02.004
Khoa DVA, Trieu LC, Cong NT, Dung TH, Thuy NTD, Quan TK, Quan DQ, Trang NTT, Linh NTN, Tuong NH, Vy PNT (2025). Genetic diversity in Vietnamese Ven dogs: A comprehensive assessment through D-loop hypervariable region 1 sequences. Aust J Agric Vet Anim Sci (AJAVAS), 1(3), 100007
https://doi.org/10.64902/ajavas.2025.100007
Kim KS, Lee SE, Jeong HW, Ha JH. 2001. The complete nucleotide sequence of the domestic dog (Canis familiaris) mitochondrial genome. Molecular Phylogenetics and Evolution, 10(2), 210–220. https://doi.org/10.1006/mpev.1998.0513
Koban E, Gökçek Saraç ÇG, Açan SC, Savolainen P, Togan I. 2009. Genetic relationship between Kangal, Akbash and other dog populations. Discrete Applied Mathematics, 157(10), 2335–2340. https://doi.org/10.1016/j.dam.2008.06.040
Nguyen TC, Ai NT, Thao NTT, Loc LT, Huy TTB, Hoat PC, Dung TH. 2019. Evaluation of genetic diversity of H’mong bobtail based on mitochondrial D-loop sequence. Journal of Animal Husbandry Sciences and Technics, 249, 17–22. https://csdlkhoahoc.hueuni.edu.vn/data/2019/11/2019_TAPCHICHANNUOI249.pdf
Okumura N, Ishiguro N, Nakano M, Matsui A, Sahara M. 1996. Intra- and interbreed genetic variations of mitochondrial DNA major non-coding regions in Japanese native dog breeds (Canis familiaris). Animal Genetics, 27(6), 397–405. https://doi.org/10.1111/j.1365-2052.1996.tb00506.x
Pang JF, Kluetsch C, Zou XJ, Zhang AB, Luo LY, Angleby H, Ardalan A, Ekström C, Sköllermo A, Lundeberg J, Matsumura S, Leitner T, Zhang YP, Savolainen P. 2009. mtDNA data indicate a single origin for dogs South of Yangtze River, less than 16,300 years ago, from numerous wolves. Molecular Biology and Evolution, 26(12), 2849–2864. https://doi.org/10.1093/molbev/msp195
Pereira L, Van Asch B, Amorim A. 2004. Standardisation of nomenclature for dog mtDNA D-loop: A prerequisite for launching a Canis familiaris database. Forensic Science International, 141(2–3), 99–108. https://doi.org/10.1016/j.forsciint.2003.12.014
Quan KT, Nguyen VT, Trinh TN, Huynh VH, Solvent CA. 2016a. Evaluation of genetic diversity of Phu Quoc ridgeback dogs based on mitochondrial DNA Hypervariable-1 region. Vietnam Journal of Biotechnology, 14(1A), 245–253. https://scholar.google.com/citations?view_op=view_citation&hl=en&user=PkOfpmYAAAAJ&citation_for_view=PkOfpmYAAAAJ:qjMakFHDy7sC
Quan KT, Huynh VH, Chung AD, Tran HD. 2016b. Evaluation of genetic diversity of Vietnamese dogs based on mitochondrial DNA hypervariable-1 region. Научный результат. Серия «Физиология», 23(9), 45–50. https://scholar.google.com/citations?view_op=view_citation&hl=en&user=PkOfpmYAAAAJ&citation_for_view=PkOfpmYAAAAJ:qjMakFHDy7sC
Quan KT, Nguyen VT, Huynh VH, Nguyen TC, Tran HD. 2016c. Develop a procedure for extracting DNA from dog hair. Academia Journal of Biology, 38(1), 124–132. https://doi.org/10.15625/0866-7160/v38n1.7056
Ren Z, Chen H, Yang X, Zhang C. 2017. Phylogenetic analysis of Tibetan mastiffs based on mitochondrial hypervariable region I. Journal of Genetics, 96, 119–125 (2017). https://doi.org/10.1007/s12041-017-0753-3
Rozas J, Ferrer-Mata A, Sánchez-DelBarrio JC, Guirao-Rico S, Librado P, Ramos-Onsins SE, Sánchez-Gracia A. 2017. DnaSP 6: DNA sequence polymorphism analysis of large data sets. Molecular Biology and Evolution, 34(12), 3299–3302. https://doi.org/10.1093/molbev/msx248
Savolainen P, Zhang YP, Luo J, Lundeberg J, Leitner T. 2002. Genetic evidence for an East Asian origin of domestic dogs. Science, 298(5598), 1610–1613. https://doi.org/10.1126/science.1073906
Thai QK, Chung DA, Tran HD. 2017. Canis mtDNA HV1 database: a web-based tool for collecting and surveying Canis mtDNA HV1 haplotype in public database. BMC Genetics, 18(1), 1–7. https://doi.org/10.1186/s12863-017-0528-0
Takahasi S, Miyahara K, Ishikawa H, Ishiguro N, Suzuki M. 2002. Lineage classification of canine inheritable disorders using mitochondrial DNA haplotypes. Journal of Veterinary Medical Science, 64(3), 255–259. https://doi.org/10.1292/jvms.64.255
Tamura K, Stecher G, Kumar S. 2021. MEGA11: Molecular Evolutionary Genetics Analysis Version 11. Molecular Biology and Evolution, 38(7), 3022–3027. https://doi.org/10.1093/molbev/msab120
Tran HD, Quan KT, Nguyen TC, Huynh VH, Chung AD. 2016. Identification of Phu Quoc dog origin by the D-loop region sequence in the mitochondrial genome. Academia Journal of Biology, 38(2), 269–278. https://doi.org/10.15625/0866-7160/v38n2.8346
Trieu LC, Nghi CH, Khoa DVA. 2018. Some characteristics of Ven dogs in Ca Mau province. Journal of Animal Husbandry Sciences and Technics, 232, 35–39 (in Vietnamese). Cited in: Nguyen Thi Dieu Thuy, Le Cong Trieu, Huynh Thi Phuong Loan, Bui Thi Tra Mi, Nguyen Huy Tuong, DoVo Anh Khoa 2024. Analysis of genetic diversity of Ven dog breed based on microsatellite markers. Academia Journal of Biology 46(2), 93–100. https://doi.org/10.15625/2615-9023/20446
Trieu LC, Giang NT, Binh LT, Phuoc NNT, Khoa DVA. 2019. Basic indicators in the blood formula of Ven dogs. Journal of Animal Husbandry Sciences and Technics, 252, 15–20 (in Vietnamese). Cited in: Nguyen Thi Dieu Thuy, Le Cong Trieu, Huynh Thi Phuong Loan, Bui Thi Tra Mi, Nguyen Huy Tuong, DoVo Anh Khoa 2024. Analysis of genetic diversity of Ven dog breed based on microsatellite markers. Academia Journal of Biology 46(2), 93–100. https://doi.org/10.15625/2615-9023/20446
Trieu LC, Giang NT, Binh LT, Phuc PTH, Trang PT, Linh NTN, Khoa DVA. 2020. Characteristics of some measurements of Ven dogs. Journal of Animal Husbandry Sciences and Technics, 256, 19–25 (in Vietnamese). Cited in: Nguyen Thi Dieu Thuy, Le Cong Trieu, Huynh Thi Phuong Loan, Bui Thi Tra Mi, Nguyen Huy Tuong, DoVo Anh Khoa 2024. Analysis of genetic diversity of Ven dog breed based on microsatellite markers. Academia Journal of Biology 46(2), 93–100. https://doi.org/10.15625/2615-9023/20446
Van Asch B, Pereira L, Pereira F, Santa-Rita P, Lima M, Amorim A. 2005. mtDNA diversity among four Portuguese autochthonous dog breeds: a fine-scale characterisation. BMC Genetics, 6, 37. https://doi.org/10.1186/1471-2156-6-37.
Disclaimer/Publisher’s Note: The statements, opinions, institutional affiliations, data contained in all publications, and all responsibilities for accuracy are solely those of the individual author(s) and contributor(s) and not of MARCIAS AUSTRALIA and AJAVAS/or the Editor(s). MARCIAS AUSTRALIA and AJAVAS/or the Editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content.